Practical 2: Analysis of RNA-seq data
- Introduction
- Set up your working environment
- Sequence quality controls
- Mapping of reads on the reference genome
- Post-processing of alignment files
- Alignments visualization using a genome browser
- Gene counts estimation
- Search for differentially expressed genes
Introduction
Objective of this practical session
During this practical session, you will learn:
- To conduct an analysis of RNA-seq raw fastq files. You will first perform quality control of the sequencer file outputs before proceding to the mapping of the reads onto the reference genome.
- To perform statistical analysis of the gene expression matrices in order to identify differentialy expressed genes between two conditions.
Dataset
Data used in these practical were collected from the following publication: Guida, A., Lindstädt, C., Maguire, S. L., Ding, C., Higgins, D. G., Corton, N. J., Berriman, M., et al. (2011). Using RNA-seq to determine the transcriptional landscape and the hypoxic response of the pathogenic yeast Candida parapsilosis. Guida et al. BMC Genomics 2011
The raw sequencing files located into the projet data folder of the IFB-core Cluster : /shared/projects/ens_hts_2021/ where retrieved from the EBI European Electronic Archive using the study accession number PRJNA154483.
Set up your working environment
-
Connect to the IFB-core server. Look at the tutorial to see how to proceed. You can also look at the useful commands to work on the IFB-core Cluster page.
-
Go to your home directory
cd -
Create a directory that will contain all results of the upcoming analyses
mkdir RNAseq -
Go to the newly created directory
cd RNAseq -
Check your are in the right directory using the
pwdcommand:pwd /shared/home/<your login>/RNAseq
Sequence quality controls
Fastq files are raw results from RNA-seq experiments. These files comprise all the sequences (or reads) obtained from the sequencer device (Illumina technology in this practical), together with base quality scores (PHRED scores).
Two different files will be analyzed during this practical from Guida et al. publication:
- O2rep2_SRR352263.fastq refereed to a transcriptome analysis of yeasts C. parapsilosis under normoxic condition (ENA access number: SRR352263)
- noO2rep3_SRR352271.fastq refereed to a transcriptome analysis of yeasts C. parapsilosis under hypoxic condition (ENA access number: SRR352271)
In a first step, controls will be performed on each FASTQ files in order to evaluate the overall quality of the sequences and identify potential problems that could impact the following analyses. During this practical we will use the FastQC software solution.
FastQC is a quality control application for high throughput sequence data. It provides an interactive application to review the results of several different quality control checks, and create an HTML based report. The main functions of FastQC are:
- Import of data from BAM, SAM or FastQ files (any variant)
- Providing a quick overview to tell you in which areas there may be problems
- Summary graphs and tables to quickly assess your data
- Export of results to an HTML based report
- Offline operation mode to allow automated generation of reports without running the interactive application
What you have to do:
- Use FASTQC to evaluate the quality of sequences in each FASTQ files. Using information from the Fastqc help page as well as exemples of good or bad illumina data as references.
- Compare results between the two FASTQ files. Is there any concern related to the following analyses?
-
Create a new directory to store the output of fastqc
mkdir 1-QC # Using the `tree` command, your directory should look like this: tree │ └───1-QC -
Go to this directory
cd 1-QC -
Make fastqc available in your environment
module load fastqc/0.11.9 -
Check the help page of the programme to see its usage and parameters
srun fastqc --help -
Run fastqc on each experiment files
# O2 condition # Absolute path to the file: /shared/projects/ens_hts_2021/data/rnaseq/O2rep2_SRR352263.fastq # -o option creates all output files in the specified output directory, '.' means current directory srun fastqc /shared/projects/ens_hts_2021/data/rnaseq/O2rep2_SRR352263.fastq -o . # noO2 condition srun fastqc /shared/projects/ens_hts_2021/data/rnaseq/noO2rep3_SRR352271.fastq -o .At this point you should see the new files in your directory using the
treecommandtree │ └───1-QC └─── O2rep2_SRR352263.fastqc.html └─── O2rep2_SRR352263.fastqc.zip └─── noO2rep3_SRR352271.fastqc.html └─── noO2rep3_SRR352271.fastqc.zip -
Download the HTML file reports on your local machine
## OPEN A NEW TERMINAL # Create a directory where to put generated files on your computer mkdir ~/Desktop/RNAseq/ # Go to this directory cd ~/Desktop/RNAseq/ # Download html report files from IFB server scp <login>@core.cluster.france-bioinformatique.fr:~/RNAseq/1-QC/*.html . # Enter your password -
Open the .html report with your internet browser
Mapping of reads on the reference genome
Once data quality is verified, reads will be mapped onto the reference genome of yeast C. parapsilosis. The genome sequence of C. parapsilosis and its annotations (locations of ORFs) were retrieved from the Candidat genome database (CGD).
Different aligner and algorithms for RNA-seq analysis exist. We will use Bowtie 1.2 an ultrafast (memory-efficient) short read aligner. As an input, Bowtie uses a Fastq file (with reads to be aligned) and “pre-built indexes” of the reference genome. These indexes are named “C_parapsilosis.1.ebwt”, “C_parapsilosis.2.ebwt”, etc. They will allow boosting the alignment process.
As an output, Bowtie provides a SAM file. SAM (Sequence Alignment/Map) is a generic format for storing large nucleotide sequence alignments.
What you have to do:
- Run sequence alignments with Bowtie using the two Fastq files
- While Bowtie is running, take a look at Bowtie documentation that describes the options (particularly the -m)
- Look at the alignment statistic outputs (file .out)
- What is the proportion of reads aligned on the reference genome?
- Is there any concern to take into account for the following analyses?
-
Create a new directory to store the output of bowtie
#Go back to the parental directory "RNAseq" cd ../ #Create a new directory to store results of the alignment mkdir 2-MappingYour directory should now look like this :
tree │ └───1-QC └─── O2rep2_SRR352263.fastqc.html └─── O2rep2_SRR352263.fastqc.zip └─── noO2rep3_SRR352271.fastqc.html └─── noO2rep3_SRR352271.fastqc.zip └─── 2-Mapping -
Go to the newly created directory
cd 2-Mapping -
Load Bowtie into your environment
module load bowtie/1.2.2 -
Map the reads to the reference genome
## We will use the following options: # "-S" will output the result in SAM format # "/shared/projects/ens_hts_2021/data/rnaseq/bowtie_indexes/C_parapsilosis" specify the location and the "prefix (C_parapsilosis)"" of the bowtie's index files # "/shared/projects/ens_hts_2021/data/rnaseq/Fastqc/O2rep2_SRR352263.fastq.gz" location of the input fastq # "2>" will save in a file some statistic about the aligment (#of reads mapped, etc...) # "> redirects the mapping output into a .sam file # Map the aerobic condition reads srun bowtie -S /shared/projects/ens_hts_2021/data/rnaseq/bowtie_indexes/C_parapsilosis /shared/projects/ens_hts_2021/data/rnaseq/O2rep2_SRR352263.fastq 2> O2rep2_SRR352263_bowtie_mapping.out > O2rep2_SRR352263_bowtie_mapping.sam # Map the hypoxic condition reads srun bowtie -S /shared/projects/ens_hts_2021/data/rnaseq/bowtie_indexes/C_parapsilosis /shared/projects/ens_hts_2021/data/rnaseq/noO2rep3_SRR352271.fastq 2> noO2rep3_SRR352271_bowtie_mapping.out > noO2rep3_SRR352271_bowtie_mapping.samYour directory should now look like this :
tree . ├── 1-QC │ ├── noO2rep3_SRR352271_fastqc.html │ ├── noO2rep3_SRR352271_fastqc.zip │ ├── O2rep2_SRR352263_fastqc.html │ └── O2rep2_SRR352263_fastqc.zip └── 2-Mapping ├── noO2rep3_SRR352271_bowtie_mapping.out ├── noO2rep3_SRR352271_bowtie_mapping.sam ├── O2rep2_SRR352263_bowtie_mapping.out └── O2rep2_SRR352263_bowtie_mapping.sam
Post-processing of alignment files
In order to facilitate alignement manipulation, SAM files have to be converted into BAM files (a binary version) and alignements “sorted” according to their localisation on the genome and files indexed in order to speed up their access. We will use the Samtools suite to perform these steps.
What you have to do:
- Convert SAM files into BAM files
- Sort and index BAM files
-
Sort and convert .sam into .bam files
## "samtools sort" sort alignments by genomic coordinates # "|" "pipe" the output of samtools sort to the next program ## "samtools view" will convert sam into bam # option "-b" specify the output to be in BAM format # ">"" write the output in the bam file module load samtools/1.9 # Sort and convert O2 condition srun samtools sort O2rep2_SRR352263_bowtie_mapping.sam | srun samtools view -b > O2rep2_SRR352263_bowtie_sorted.bam # Sort and convert noO2 condition srun samtools sort noO2rep3_SRR352271_bowtie_mapping.sam | srun samtools view -b > noO2rep3_SRR352271_bowtie_sorted.bam -
Create indexes for the bam files
The index of a bam file is name .bam.bai
# Index the O2 condition srun samtools index O2rep2_SRR352263_bowtie_sorted.bam # Index the noO2 condition srun samtools index noO2rep3_SRR352271_bowtie_sorted.bamYour directory should now look like this :
tree . ├── 1-QC │ ├── noO2rep3_SRR352271_fastqc.html │ ├── noO2rep3_SRR352271_fastqc.zip │ ├── O2rep2_SRR352263_fastqc.html │ └── O2rep2_SRR352263_fastqc.zip └── 2-Mapping ├── noO2rep3_SRR352271_bowtie_mapping.out ├── noO2rep3_SRR352271_bowtie_mapping.sam ├── noO2rep3_SRR352271_bowtie_sorted.bam ├── noO2rep3_SRR352271_bowtie_sorted.bam.bai ├── O2rep2_SRR352263_bowtie_mapping.out ├── O2rep2_SRR352263_bowtie_mapping.sam ├── O2rep2_SRR352263_bowtie_sorted.bam └── O2rep2_SRR352263_bowtie_sorted.bam.bai
Alignments visualization using a genome browser
The Integrative Genomics Viewer (IGV) is a high-performance visualization tool for interactive exploration of large genomic datasets. It supports a wide variety of data types, including array-based, high throughput sequence data and genomic annotations. In this practical, we will use IGV to visualize mapping results.
-
Download the necessary files on your computer
# To download files from the cluster to your current directory (on your own computer), **open a new shell and run** # First bam and bai alignement files scp <your login>@core.cluster.france-bioinformatique.fr:~/RNAseq/2-Mapping/*.bam* . # Next the reference genome sequence and gene annotation files scp <your login>@core.cluster.france-bioinformatique.fr:/shared/projects/ens_hts_2021/data/rnaseq/C_parapsilosis_CGD.fasta . scp <your login>@core.cluster.france-bioinformatique.fr:/shared/projects/ens_hts_2021/data/rnaseq/C_parapsilosis_ORFs.gff . -
Visualize mapping results with IGV
On linux type “igv” in a terminal window to launch the program
You can also download IGV and follow the instalation instructions according to your OS
Once the IGV program is launched, it is necessary to import the reference genome “Genomes/Create .genome File…” (see below). Select the FASTA file with the genomic sequence of C. parapsilosis “C_parapsilosis_CGD.fasta” (“Browse / FASTA file”) and enter information regarding ORFs positions, GFF file “C_parapsilosis_ORFs.gff” (“Browse Gene file”).
Finally, give a name to your genome (“Unique identifier” and “Descriptive name”) and press “OK”. Save the genome file in your home.Warning! In order to IGV to create an index of your genome, you need to copy the reference genome FASTA file in writable directory.
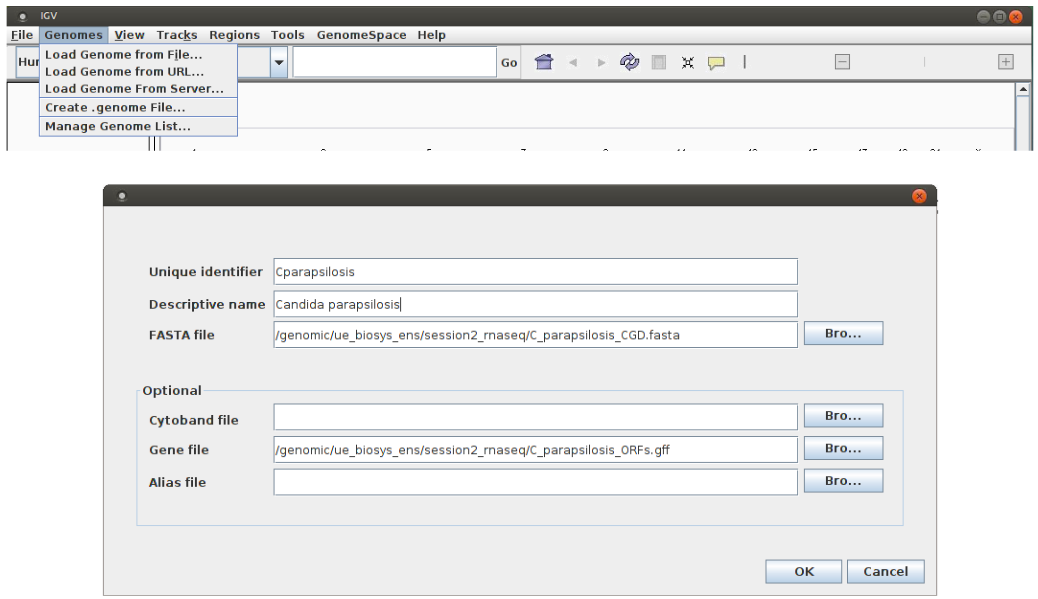
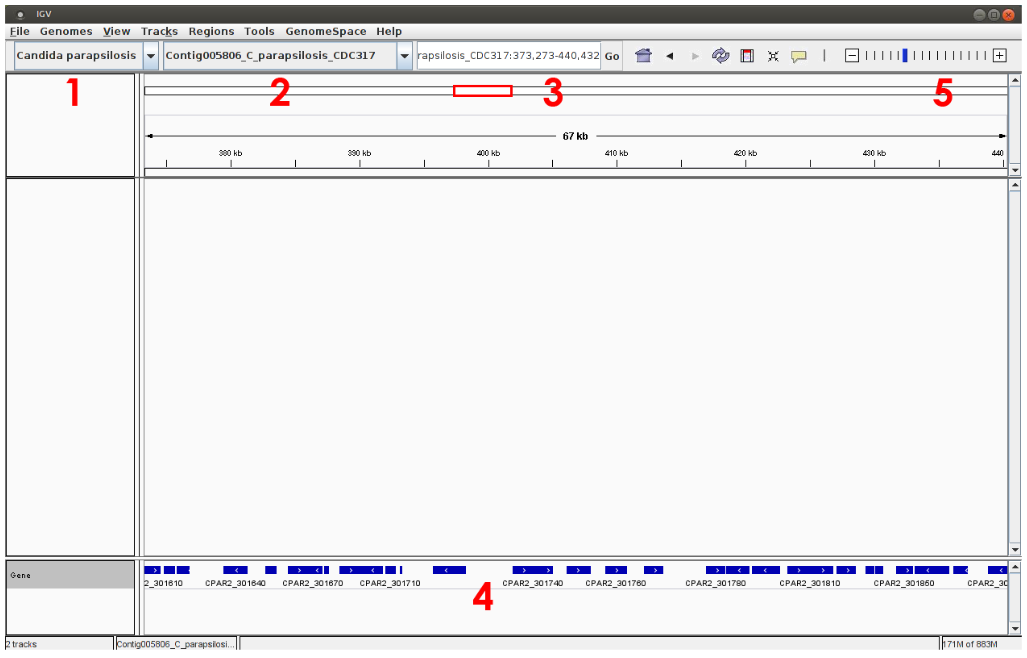
C. parapsilosis genome is now loaded into IGV and can be selected from the top/left menu (see 1 below). The genomic sequence can be therefore explored, choosing for instance, a particular chromosome (see 2 below) or a genomic region (see 3). Note that gene annotations (ORF positions) are shown at the bottom of the window (see 4, blue lines) and you can obtain a more detailed view of the sequence using the cursor located on the top/right of the window, see 5).
Mapping results (“.sorted.bam” files) can now be imported (“File / Load from File”). Zoom in genomic regions in order to visualize read alignements.
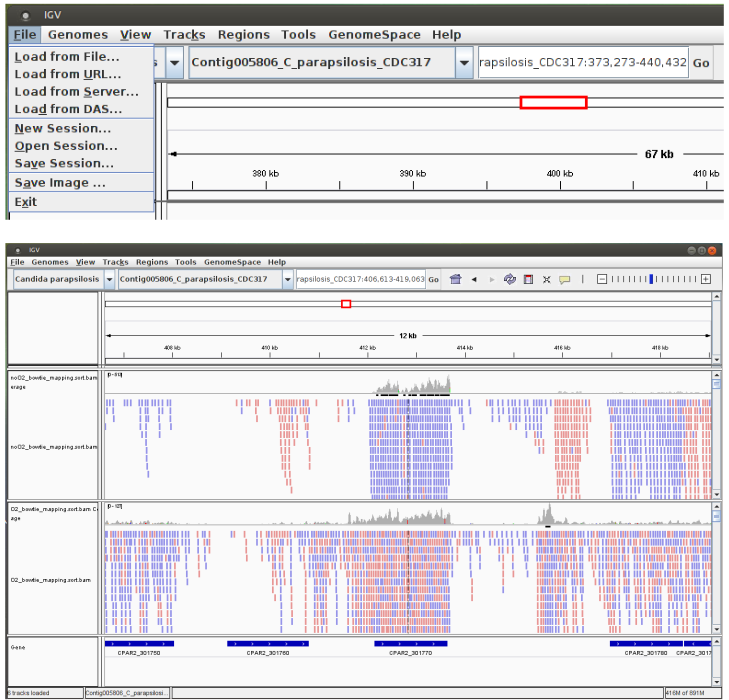
What you have to do:
- Compare your results with those presented in the original publication
- Did the authors use stranded-specific protocols?
- Can you observe differences between hypoxic and normoxic conditions?
Gene counts estimation
To identify genes whose expression is different between hypoxic and normoxic conditions, we will count and compare the number of reads mapped to each ORF. A program available in the Bedtools suite will be used for this purpose.
What you have to do:
- Calculate for each ORF the number of reads that were aligned (normoxic and hypoxic conditions).
-
Create a new directory to store the ORF count matrix
# Go to the parental directory "RNAseq" cd ../ # Create a new directory to store results of the alignment mkdir 3-CountsYour directory should now look like this :
tree . ├── 1-QC │ ├── noO2rep3_SRR352271_fastqc.html │ ├── noO2rep3_SRR352271_fastqc.zip │ ├── O2rep2_SRR352263_fastqc.html │ └── O2rep2_SRR352263_fastqc.zip ├── 2-Mapping │ ├── noO2rep3_SRR352271_bowtie_mapping.out │ ├── noO2rep3_SRR352271_bowtie_mapping.sam │ ├── noO2rep3_SRR352271_bowtie_sorted.bam │ ├── noO2rep3_SRR352271_bowtie_sorted.bam.bai │ ├── O2rep2_SRR352263_bowtie_mapping.out │ ├── O2rep2_SRR352263_bowtie_mapping.sam │ ├── O2rep2_SRR352263_bowtie_sorted.bam │ └── O2rep2_SRR352263_bowtie_sorted.bam.bai └── 3-Counts -
Go to the newly created directory
cd 3-Counts -
Calculate for each ORF the number of reads that were aligned to it
module load bedtools/2.27.1 # Counting matrix for the O2 condition srun bedtools multicov -bams ../2-Mapping/O2rep2_SRR352263_bowtie_sorted.bam -bed /shared/projects/ens_hts_2021/data/rnaseq/C_parapsilosis_ORFs.gff > O2rep2_SRR352263_gene_counts.gff # Output formating srun sed 's/^.*ID=//' O2rep2_SRR352263_gene_counts.gff > O2rep2_SRR352263_gene_counts.tab # Counting matrix for the noO2 condition srun bedtools multicov -bams ../2-Mapping/noO2rep3_SRR352271_bowtie_sorted.bam -bed /shared/projects/ens_hts_2021/data/rnaseq/C_parapsilosis_ORFs.gff > noO2rep3_SRR352271_gene_counts.gff # Output formating srun sed 's/^.*ID=//' noO2rep3_SRR352271_gene_counts.gff > noO2rep3_SRR352271_gene_counts.tabTake a look at the final counting matrices to see how the files are organised.
At the end of RNA-seq data analysis your directory should look like this :
tree . ├── 1-QC │ ├── noO2rep3_SRR352271_fastqc.html │ ├── noO2rep3_SRR352271_fastqc.zip │ ├── O2rep2_SRR352263_fastqc.html │ └── O2rep2_SRR352263_fastqc.zip ├── 2-Mapping │ ├── noO2rep3_SRR352271_bowtie_mapping.out │ ├── noO2rep3_SRR352271_bowtie_mapping.sam │ ├── noO2rep3_SRR352271_bowtie_sorted.bam │ ├── noO2rep3_SRR352271_bowtie_sorted.bam.bai │ ├── O2rep2_SRR352263_bowtie_mapping.out │ ├── O2rep2_SRR352263_bowtie_mapping.sam │ ├── O2rep2_SRR352263_bowtie_sorted.bam │ └── O2rep2_SRR352263_bowtie_sorted.bam.bai └── 3-Counts ├── noO2rep3_SRR352271_gene_counts.gff ├── noO2rep3_SRR352271_gene_counts.tab ├── O2rep2_SRR352263_gene_counts.gff └── O2rep2_SRR352263_gene_counts.tab -
Unload the tools you used
module unload fastqc/0.11.9 bowtie/1.2.2 samtools/1.9 bedtools/2.27.1
Search for differentially expressed genes
In their article (Guida et al., 2011), the authors repeated the experiment 6 times for normoxic condition (with O2) and 4 times for hypoxic conditions (without O2). Results obtained for all experiments are combined in the file “rnaseqcount_data_diffAnalysis.txt”. This file will be used to search for differentially expressed genes using the DESeq2 (Love et al. 2014) method.
The DESeq package provides methods to test for differential expression by use of the negative binonial distribution and a shrinkage estimator for the distribution’s variance.
What you have to do:
- Search for differentially expressed genes using DESeq R package
- How many genes are selected with different adjusted p-value thresholds (5%, 1%, etc.)?
- Check your results with IGV and use GOtermFinder (see practical on microarrays) to analyse the function of the selected genes
-
Connect to Rstudio server of the IFB Look at the tutorial on how to connect to IFB-core Rstudio server to see how to proceed.
-
Save the working notebook in your personal environment
- In File > Open File… enter the path /shared/projects/ens_hts_2021/data/rnaseq/DEseq2.Rmd to open the notebook containing all the code needed for the practical
- Save it into your personal folder using File > Save As
-
Follow the instruction of the notebook to conduct the analysis. You can also visualize the final report version.
You can find help on how to use R markdown on the R markdown project webpage.